Chemical modification
Modifications can be added to oligonucleotides either during the synthesis or post-synthetically (after having finished the automated synthesis and cleavage from the support). Several methods are possible. Their usage depends on the availability of reagents and the position where the modification should be added to the oligo – 5’, 3’ or internal. The chemical modification of oligonucleotides is more efficient, giving higher product yields than protocols based on enzymatic approaches.
Phosphoramidites
Phosphoramidites
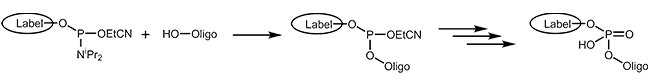
The easiest and fastest method to add a modification to an oligonucleotide – at the 5’-end or internally – is coupling it as a phosphoramidite during synthesis. Basic to this method is that the modification is available as a phosphoramidite building block. In addition, the modification must be stable to all chemical reagents used during the further steps, in particular during the cleavage from the support and the removal of the protecting groups.
A variety of dyes and labels (e.g. FAM, HEX, TET, Biotin) as well as linkers and reactive groups (e.g. Amino, Thiol) that are used for further modification, are incorporated this way.
 |
NHS-Ester and aminolink
NHS-Ester and aminolink
Many fluorescent dyes and biomolecules can be coupled as reactive N-hydroxy-succinimide-esters to oligonucleotides carrying a primary amino function (Aminolink). The NHS-ester is reacted with the oligo in organic-aqueous solution in a post-synthetic reaction step. As a result the modification is covalently linked to the oligo by an amide bond. As this type of reaction is not quantitative, a careful purification of product by HPLC is always necessary.
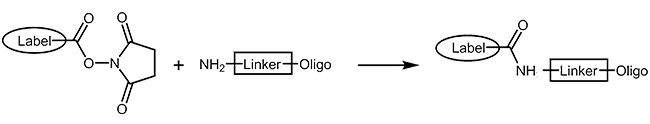 |
Thiol and maleimide
Thiol and maleimide
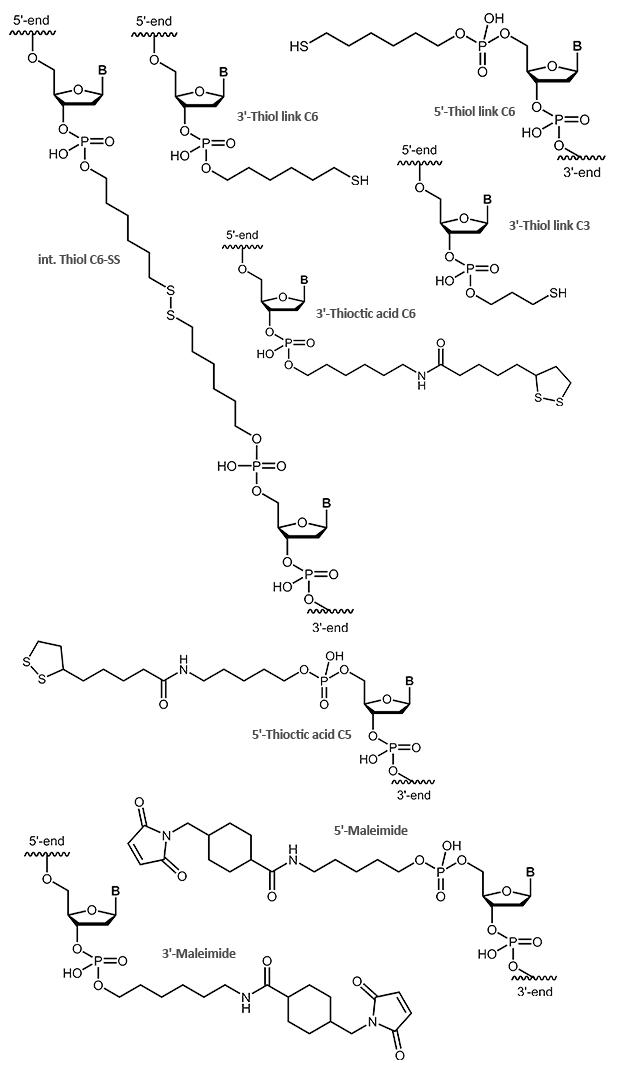
Thiol and maleimides are easily reacted forming a stable covalent linkage with each other. Dependent on the reactive agent available for the modifier, the oligo can either be activated with a thiol or a maleimide function, both options are available.
|
|
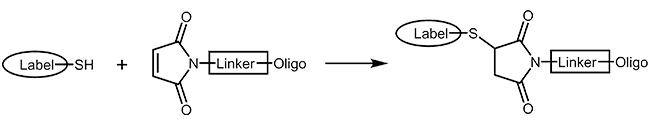
Click chemistry
Click chemistry
The term „Click Chemistry“ describes a fast and thermodynamically favoured reaction which enables an efficient and selective linkage of two molecules. In a more specific sense the click reaction is a cycloaddition between an azide and an alkyne either under copper catalyzed or copper free reaction conditions. Due to the fact that such click reactions work efficiently in aqueous media, they are very much suitable for modifying biomolecules or linking different biomolecules together. Furthermore the azide and alkyne reaction partners do not interfere with other functional groups like e.g. amino or carboxy which opens an additional degree of freedom for orthogonal coupling strategies.
| Copper catalyzed azide alkyne cycloaddition |
|---|
|
The copper catalyzed azide alkyne cycloaddition (CuAAC)1,2 is an improvement of the classical Huisgen cycloaddition3. In both cases a terminal alkyne reacts with an azide to form a 1,2,3-triazole. In comparison to the original Huisgen reaction, which is non-regioselective and only works at elevated temperature, the copper catalyzed version is several times faster, runs efficiently at room temperature and selectively gives the 1,4-regioisomer of the triazole. |
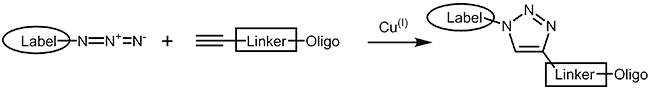 |
| Copper free click reaction |
|---|
|
As the copper catalyst is toxic for bacteria and mammalian cells, a copper-free chemistry has been developed that also runs under physiological conditions. A short review of Becer et al.4 gives a good survey on click chemistry reactions that work as fast and straightforward as the CuAAC but without involving any metal ions. Cyclooctynes5 with several substituent patterns are preferred compounds for strain promoted azide alkyne cycloadditions. |
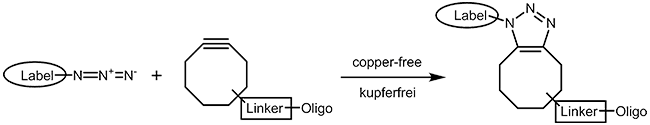 |
Literature:
1. Tornøe CW, Christensen C, Meldal MJ. 2002. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific Copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. Org. Chem. 67, 3057-3064.
2. Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. 2002. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 41, 2596-2599.
3. Huisgen R. 1961. 1,3-Dipolar cycloadditions. Proc. Chem. Soc. 357-396.
4. Becer CR, Hoogenboom R, Schubert US. 2009. Click chemistry beyond metal-catalyzed cycloaddition. Angew. Chem. Int. Ed. 48, 2-11
5. Agard NJ, Prescher JA, Bertozzi CR. 2004. A Strain-promoted [3 + 2] Azide−alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem.Soc. 126, 15046-15047

